Галактоземия у детей. Клинические рекомендации.
Галактоземия у детей
Оглавление
Ключевые слова
Тотальная (общая) галактоза
Список сокращений
ГАЛК – фермент галактокиназа
ГАЛТ – фермент галактозо-1-фосфатуридилтраснфераза
ГАЛЭ – фермент УДФ-галактозо-4-эпимераза (эпимераза)
КТ – компьютерная томография
МРТ – магнитно-резонансная томография
УЗИ – ультразвуковое исследование
Термины и определения
Неонатальный скрининг – медицинская диагностическая технология сплошного безвыборочного лабораторного обследования всех новорожденных на некоторые заболевания обмена веществ, призванная обеспечить своевременное выявление и начало лечения больных детей с целью предотвращения их инвалидизации.
Овариальная недостаточность – патологическое состояние, которое проявляется исходным отсутствием, снижением или преждевременным прекращением овуляторной и секреторной функции яичников.
Пренатальная диагностика галактоземии – комплексная дородовая диагностика с целью выявления галактоземии на стадии внутриутробного развития путем определения активности галактозо-1-фосфатуридилтрансферазы (ГАЛТ) в культуре амниоцитов, биоптате и культуре хориона.
1. Краткая информация
1.1 Определение
1.2 Этиология и патогенез
Галактоземия относится к наследственным болезням углеводного обмена и объединяет несколько генетически гетерогенных форм. В основе заболевания лежит недостаточность одного из трех ферментов, участвующих в метаболизме галактозы: галактозо-1-фосфатуридилтраснферазы (ГАЛТ), галактокиназы (ГАЛК) и уридин-дифосфат(УДФ)-галактозо-4-эпимиразы (ГАЛЭ) (рисунок 1). Известны три гена, мутации в которых могут приводить к развитию галактоземии (таблица 1).
УДФ-галактозо-4-эпимераза (ГАЛЭ, эпимераза)
Галактоза играет крайне важную роль в организме, особенно растущего ребенка. Этот моносахарид не только является значимым источником энергии для клетки, но и служит необходимым пластическим материалом для образования гликопротеидов, гликолипидов и других комплексных соединений, используемых организмом для формирования клеточных мембран, нервной ткани, нервных окончаний, процессов миелинизации нейронов и др. Нарушение метаболизма галактозы, наблюдаемое при галактоземии, неизбежно приводит к расстройству функционирования многих органов и систем организма.
Основным источником галактозы у человека является пища. Большое количество потребляемых в течение дня пищевых продуктов содержат лактозу, из которой в кишечнике в результате гидролиза образуется галактоза; многие продукты питания содержат галактозу в чистом виде. У человека галактоза может образовываться эндогенным путем, подавляющее ее количество синтезируется в процессе ферментативных реакций между уридиндифосфатглюкозой (УДФ-глюкозой) и УДФ-галактозой, а также в процессе обмена гликопротеинов и гликолипидов.
Патогенетические механизмы галактоземии до сих пор полностью не изучены. В результате недостаточности любого из трех ферментов – ГАЛТ, ГАЛК или ГАЛЭ – в крови повышается концентрация галактозы. При дефиците активности ферментов ГАЛТ и ГАЛЭ, помимо избытка галактозы, в организме больного накапливается также избыточное количество галактозо-1-фосфата, что на сегодняшний день считается основным патогенетическим фактором, обусловливающим большинство клинических проявлений галактоземии и формирование отсроченных осложнений. Избыток галактозы в организме может метаболизироваться другими биохимическими путями: в присутствии НАДФ?Н (или НАД?Н) она может превращаться в галактитол. Накопление галактитола в крови и тканях и повышение его экскреции с мочой наблюдается при всех формах галактоземии; в хрусталике глаза избыток галактитола способствует формированию катаракты. Имеются сведения о том, что высокое содержание галактитола в тканях мозга способствует набуханию нервных клеток и формированию псевдоопухоли мозга у отдельных больных.
Патологические процессы при галактоземии обусловлены не только токсическим действием указанных продуктов, но и их тормозящим влиянием на активность других ферментов, участвующих в углеводном обмене (фосфоглюкомутазы, глюкозо-6-фосфатдегидрогеназы), следствием чего является гипогликемический синдром.
Предполагается также, что предрасположенность к сепсису у новорожденных с галактоземией обусловлена ингибированием бактериальной активности лейкоцитов.
1.3 Эпидемиология
Частота галактоземии по данным массового обследования новорожденных в России составляет 1: 20 000, при этом подавляющее большинство случаев заболевания обусловлено мутациями в гене GALT.
1.4 Кодирование по МКБ-10
1.5 Примеры диагнозов
Классическая галактоземия тип I
Недостаточность уридиндифосфат-галактозо-4-эпимеразы (ГАЛЭ или эпимеразы) – галактоземия тип III
Нарушения обмена галактозы
1.6 Классификация
В основу современной классификации галактоземии положен этиологический принцип. Существуют три типа галактоземии, в зависимости от имеющегося у больного дефекта одного из трех основных ферментов, участвующих в метаболизме галактозы:
2. Диагностика
2.1 Жалобы и анамнез
На фоне вскармливания молоком у новорожденного появляется рвота, диарея, мышечная гипотония, сонливость, вялость. Останавливается прибавка в массе тела, наблюдается вялое сосание, отказ от груди матери, появляются и нарастают признаки поражения печени, часто сопровождающиеся гипогликемией, желтухой и гепатоспленомегалией, нередко отмечается кровоточивость из мест инъекций.
2.2 Физикальное обследование
Рекомендуется проведение стандартного подробного физикального осмотра пациента с подозрением на галактоземию [1,6].
(Сила рекомендации A; уровень убедительности доказательств I)
Комментарий: Могут быть выявлены: снижение массы тела, иктеричность или бледность кожных покровов, гепатоспленомегалия, геморрагический синдром.
2.3 Лабораторная диагностика
Диагностика направлена на определение содержания в сухом пятне крови концентрации тотальной (общей) галактозы путем неонатального скрининга, при положительном результате – определении активности фермента ГАЛТ и мажорных мутации в гене ГАЛТ.
Рекомендуется проведение неонатального скрининга [1,8,10].
(Сила рекомендации A; уровень убедительности доказательств I)
При помощи флуоресцентного метода в пятнах высушенной крови проводят определение уровня тотальной галактозы, который представляет собой сумму концентрации галактозы и галактозо-1-фосфата (таблица 2).
Уровень тотальной галактозы в сыворотке крови (мг/дл)
При выявлении уровня тотальной галактозы 7,2 мг/дл и выше рекомендуется проведение подтверждающей диагностики классической галактоземии, которая включает в себя определение активности фермента ГАЛТ и ДНК-диагностику с целью выявления наиболее распространенных мутаций в гене GALT и полиморфного варианта Дуарте, приводящего к снижению активности фермента [3,4,5,12].
(Сила рекомендации A; уровень убедительности доказательств I)
Комментарий: Определение активности ГАЛТ проводят в пятнах высушенной крови и/или в цельной гепаринизированной крови, ДНК-исследование осуществляют в образце цельной крови. Взятие образца крови для ДНК-исследования производят из периферической вены с добавлением в пробирку консерванта – ЭДТА или гепарина.
Диагностически значимым считается снижение активности фермента ГАЛТ в высушенных пятнах крови c, p.Met142Lys, p.Leu358Pro, составляющие в совокупности 82,1% мутантных аллелей в российской популяции, и p.Asn314Asp (N314D, вариант Дуарте). Алгоритм диагностики классической галактоземии представлен на рисунке 1.
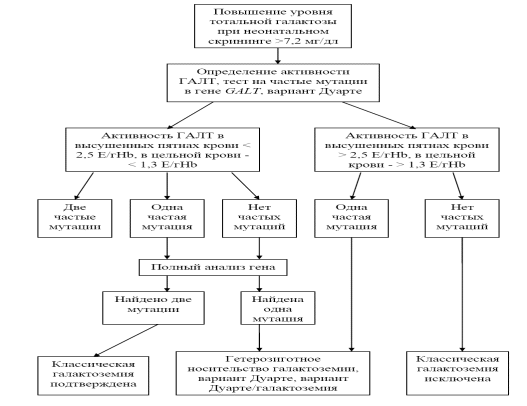
Рисунок 1. Алгоритм подтверждающей диагностики галактоземии I типа.
2.4 Инструментальная диагностика
Рекомендуется проведение ультразвукового исследования (УЗИ) органов брюшной полости и почек для выявления гепато-, сплено- и нефромегалии, асцита [1, 6].
(Сила рекомендации C; уровень убедительности доказательств II)
Рекомендуется проведение электрокардиографии (ЭКГ) и эхокардиографии (ЭхоКГ) для обнаружения малых аномалий развития сердца [1, 6].
(Сила рекомендации C; уровень убедительности доказательств II)
Рекомендуется проведение офтальмоскопии с использованием oелевой лампы для выявления катаракты и/или другой патологии зрительного анализатора [1, 6].
(Сила рекомендации C; уровень убедительности доказательств II)
Рекомендуется проведение компьютерной томографии (КТ) / магнитно-резонансной томографии (МРТ) для диагностики органической патологии центральной нервной системы [1, 6].
(Сила рекомендации C; уровень убедительности доказательств II)
Рекомендуется проведение рентгенографии (денситометрии) для диагностики остеопороза [1, 6].
(Сила рекомендации C; уровень убедительности доказательств II)
2.5 Иная диагностика
Рекомендуются консультации специалистов (по показаниям) генетика, офтальмолога, невропатолога, диетолога, кардиолога, логопеда-дефектолога, ортопеда пациентам с галактоземией, имеющим нарушения функций соответствующих органов и систем [1, 6].
(Сила рекомендации C; уровень убедительности доказательств II)
2.6 Дифференциальная диагностика
При наличии у ребенка клинических проявлений классической галактоземии в сочетании с положительным результатом неонатального скрининга на галактоземию и нормальной активностью фермента ГАЛТ проводится поиск мутаций в гене GALE, кодирующем фермент уридиндифосфат-галактозо-4-эпимеразу (ГАЛЭ), для исключения галактоземии III типа. Далее дифференциальная диагностика проводится с:
болезнями, сопровождающимися повышенным выделением сахара (меллитуриями, сахарным диабетом, цистинозом, синдром Фанкони);
ранзиторной гипергалактоземией, которая отмечается при позднем закрытии венозного протока (Ductus venosus) и исчезает в течение 2-3-5 месяцев после его закрытия, а также у гетерозиготных носителей мажорных мутаций в генах GALT и GALE.
Поэтому для дифференциальной диагностики требуется определение в сыворотке крови: содержания общей галактозы, общего белка, альбумина, глюкозы, факторов свертывания, электролитов, С-реактивного белка, прокальцитонина, активности трансаминаз, концентрации билирубина и его фракций, альфа-фетопротеина и уровня желчных кислот, кислотно-щелочного состояния, определение содержания аминокислот. органических кислот, ацилкарнитинов и аммиака в плазме крови., биохимический анализ мочи [1,7].
3. Лечение
3.1 Консервативное лечение
Целью лечения является полное исключение поступления в организм галактозы с пищей.
Рекомендуется элиминационная диетотерапия, предусматривающая пожизненное исключение из рациона продуктов, содержащих галактозу и лактозу [1, 2].
(Сила рекомендации A; уровень убедительности доказательств I)
Комментарии: необходимо полностью исключить из рациона больного любой вид молока (в том числе женское, коровье, козье, детские молочные смеси и др.) и все молочные продукты, а также строго избегать употребления тех продуктов, куда они могут добавляться (хлеб, выпечка, сосиски, колбасы, карамель, сладости, маргарины и т.п.). Запрещается также использование низколактозных смесей и молока.
— печень, почки, мозги и другие субпродукты;
— печеночный паштет, ливерная колбаса;
При составлении лечебных рационов для детей первого года жизни количество основных пищевых ингредиентов и энергии должно быть приближено к физиологическим нормам.
Для лечения больных галактоземией используются специализированные смеси на основе изолята соевого белка, гидролизатов казеина, безлактозные казеинпредоминантные молочные смеси, а также смеси на основе синтетических аминокислот.
Рекомендуется в качестве лечебного продукта использовать смеси на основе изолята соевого белка, в которых полностью отсутствуют растительные галактозиды [1,8,10].
(Сила рекомендации B; уровень убедительности доказательств II)
Комментарии: при использовании соевых смесей (таблица 4) в питании грудных детей возможно появление аллергических реакций на белок сои.
Галактоземия у детей
Общая информация
Краткое описание
СОЮЗ ПЕДИАТРОВ РОССИИ
Клинические рекомендации: Галактоземия у детей
Год утверждения (частота пересмотра): 2016 (пересмотр каждые 3года)

Автоматизация клиники: быстро и недорого!
— Подключено 300 клиник из 4 стран

Автоматизация клиники: быстро и недорого!
Мне интересно! Свяжитесь со мной
Классификация
Этиология и патогенез
| Фермент | Название гена | Локализация гена |
| Галактозо-1-фосфат-уридилтрансфераза (ГАЛТ) | GALT | 9p13.3 |
| Галактокиназа (ГАЛК) | GALK1 | 17q25.1 |
| УДФ-галактозо-4-эпимераза (ГАЛЭ, эпимераза) | GALE | 1р36.11 |
Эпидемиология
Частота галактоземии по данным массового обследования новорожденных в России составляет 1: 20 000, при этом подавляющее большинство случаев заболевания обусловлено мутациями в гене GALT.
Диагностика
Диагностика направлена на определение содержания в сухом пятне крови концентрации тотальной (общей) галактозы путем неонатального скрининга, при положительном результате – определении активности фермента ГАЛТ и мажорных мутаций в гене ГАЛТ.
| Результат | Уровень тотальной галактозы в сыворотке крови (мг/дл) |
| Отрицательный | |
| Пограничный (требуется повторное исследование) | 7,2-10 |
| Положительный | > 10 |

Рисунок 1. Алгоритм подтверждающей диагностики галактоземии I типа.
Дифференциальный диагноз
Лечение
В таких случаях целесообразно назначать смеси на основе гидролизатов казеина (таблица 5).
В зависимости от состояния ребенка возможно сочетанное применение соевой смеси и смеси на основе гидролизата казеина в соотношении 1:1. Возможно применение казеинпредоминантных безлактозных молочных смесей (таблица 6).
*соотношение казеина и сывороточных белков (%).
| Продукты и блюда | Возраст введения прикорма (мес) |
| Овощное пюре | 4-6 |
| Каши молочные | — |
| Каши безмолочные | 4-6 |
| Фруктовое пюре | 5-6 |
| Сок фруктовый | 5-6 |
| Творог | — |
| Мясное пюре | 6 |
| Яйцо (желток)* | после 12 месяцев |
| Рыба | 8 |
| Кефир и другие кисломолочные продукты | — |
| Сухари, печенье (обычные) | — |